Atrial Fibrillation
Atrial fibrillation (AF) is the most common cardiac arrhythmia. Hypertensive heart disease and coronary artery disease are risk factors. Patients with rheumatic heart disease have a high incidence of atrial fibrillation.
Older patients with chronic atrial fibrillation present with fibrosis of the atrial myocardium. Other conditions that cause atrial fibrillation include left atrial dilatation brought about by the effects of untreated hypertension. Cardiomyopathies and left-sided valvular diseases have the same physiologic sequelae. Patients who abuse alcohol, those who have contracted infections, and those diagnosed with hyperthyroidism may also be affected by the same pathophysiologic effects that lead to atrial fibrillation. Transient AF is also commonly observed in patients after open-heart surgery or acute MI.
Studies have shown that patients between 51 and 60 years of age have a prevalence rate for atrial fibrillation of 0.5–0.8%. This increases to 9% for patients 80–89 years old.

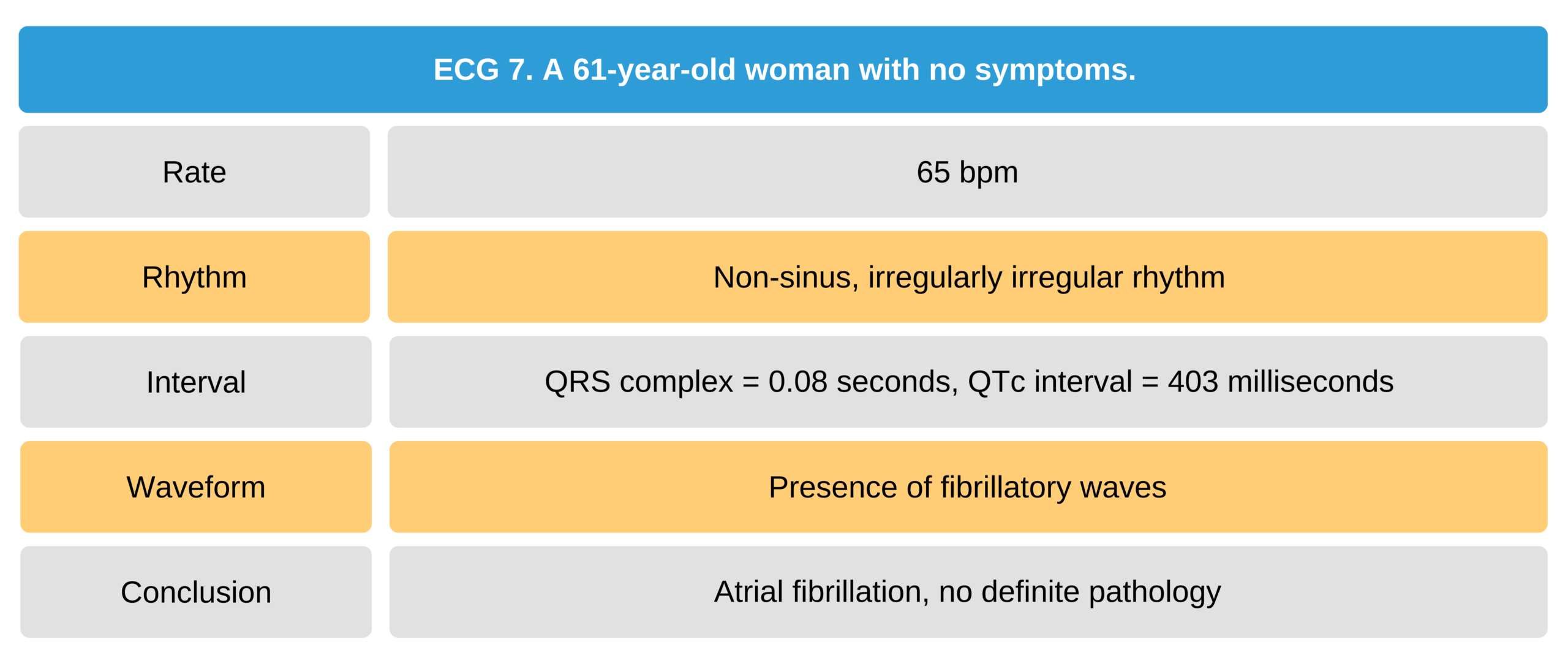
61-Year-Old Asymptomatic Woman ECG

79-Year-Old Woman With Mitral Valve Disease ECG

Related Video – ECG Rhythm Review – Atrial Fibrillation
Electrophysiology
The electrophysiologic effects of atrial fibrillation are caused by the development of complex conduction patterns and a rapid atrial rate. The normal atrial myocardium has electrophysiologic properties suitable for a short action potential duration, rapid cellular reactivation, very fast electrical conduction, and a shortened refractory period when the heart rate increases.
The “fast-response” tissues of the atrial myocardium require a rapid sodium current for phase 0 of the action potential. These same properties cause atrial fibrillation when there are anatomical changes within the atria.
Triggers
The pulmonary veins trigger the rapid atrial firing that causes atrial fibrillation. This is also known as “atrial kick.” Stretching of the atrial chamber can also elicit the rapid firing of impulses from the pulmonary veins.
Atrial fibrillation can (1) start and stop suddenly (paroxysmal), (2) last for more than 7 days (persistent), or (3) more than a year (long-standing). Triggers and substrates determine the clinical presentation of atrial fibrillation.
Substrates
Atrial remodeling occurs when there are anatomical changes in the atrium caused by fibrosis or conduction defects. Some patients exhibit both structural and conduction defects. These defects produce electrical reentrant circuits that trigger atrial fibrillation.
Early on, the relatively healthy atrium restores atrial fibrillation back to normal sinus rhythm relatively quickly. As the atrium gets remodeled over time, the return to sinus rhythm gets more difficult. In worse-case scenarios, the atrium is remodeled to the extent that the clinician can no longer correct the problem through catheter ablation therapy. In this case, the patient is diagnosed with permanent atrial fibrillation.
The autonomic nervous system is also involved in atrial fibrillation through sympathetic and parasympathetic functions. Exercise-induced atrial fibrillation is elicited by sympathetic mechanisms. Parasympathetic mechanisms are responsible for AFL in young patients with no attributable structural heart disease. Vagal stimulation may shorten atrial refractory periods and induce a substrate for reentry circuits. It also leads to focal triggers of atrial fibrillation.
Isoproterenol stimulates the sympathetic nervous system. Electrophysiology specialists use isoproterenol to induce triggers in patients undergoing catheter ablation for atrial fibrillation.
In fibrosis, the resultant structural changes are conducive for atrial fibrillation to propagate. Fibrosis intensifies the local properties of atrial cells, which includes shortening the action potential duration, increasing the dispersal of refractoriness, and shortening the conduction velocity.
Hemodynamics
The hemodynamic consequences of AFL are mild In about 50% of patients. Fortunately, some of the very rapid atrial impulses are blocked at the AV node and hindered from reaching the ventricles. This safety mechanism preserves hemodynamic stability in many patients. However, in severe cases, the fast impulses are conducted randomly to the ventricles, which leads to ventricular arrhythmias and hemodynamic consequences.
When the ventricular rate becomes fast and erratic, patients start to feel symptoms that can lead to grave hemodynamic consequences. During AFL, not all QRS complexes produce a ventricular contraction. In fact, diastole shortens, and ventricular filling becomes sub-optimal. Consequently, the ejection fraction and cardiac output become impaired.
Related Video – What is Ejection Fraction?
Related Video – What is Cardiac Output?
Clinical Significance
Patient symptoms occur when the ventricular rate exceeds 230 bpm. Symptoms may also be elicited right after spontaneous conversion of the arrhythmia to sinus rhythm because it causes a prolonged stunning of the atria and ventricles.
About half of patients diagnosed with atrial fibrillation can live a healthy life without complications or restrictions on everyday activity. Symptomatic patients experience limited work capacity, palpitations, and syncope.
When cardiac output is insufficient, the pooled blood in the ventricles increases the risk of thrombus formation, which can lead to a cerebral stroke or a pulmonary embolism. Patients with atrial fibrillation and severe heart decompensation have reduced survival rates because atrial fibrillation aggravates the already decompensated heart.
Aberrations in Atrial Fibrillation
Ashman Beats
In 1947, Gouaux and Ashman described the phenomenon of a prolonged refractory period within the right and left bundle branches with instantaneous bradycardia. This is what we know today as Ashman beats.
Ashman beats can be appreciated on longer rhythm strips. A ventricular aberration is more likely than a PVC if an abnormally long R-R interval is followed by a QRS complex that has a bundle-branch block configuration. An RBBB block aberration is more frequent than an LBBB aberration because the refractory period is more pronounced in the right bundle-branch. Ashman beats do not have a compensatory pause, but a PVC does.

Ashman Beat 35
In the ECG above (figure 5), there is a right bundle-branch block aberration (lead II) for a single beat, which occurs after a relatively long R-R interval.
Wolff-Parkinson-White Syndrome
In 1930, Louis Wolff, Sir John Parkinson, and Paul Dudley White published an article describing patients with paroxysmal tachycardias and atrial fibrillation associated with a sinus rhythm pattern of bundle-branch block and a very short PR interval. The pattern they described came to be known as Wolff-Parkinson-White Syndrome.
Wolff-Parkinson-White syndrome (WPW) is caused by anomalous aggregates of conducting cardiac tissues that bypass the atrioventricular conduction system. The accessory pathway in WPW is known as the bundle of Kent. This direct connection bypasses the AV node and leads to a preexcitation mechanism where impulses precede the usual activation of the His-Purkinje system.
The cardiac tissues in this accessory pathway are the result of a congenital malformation that developed when the myocardial syncytium was not absorbed at the annulus fibrosis of the AV valves during fetal development. This tissue conducts impulses faster than the AV node. Excitation of these tissues produces very short PR intervals.
The impulses in the accessory pathway can flow in an antegrade or retrograde manner between the atria and ventricles. Impulses travel bidrectionally in 60–75% of accessory pathways.
When an accessory pathway conducts purely in a retrograde manner, there are no delta waves produced in the WPW pattern on the ECG, but the effects of reentrant tachycardia are still evident. When an accessory pathway is conducted purely in the antegrade direction, the ECG shows a delta wave, and the WPW pattern is evident.
Atrial fibrillation and ventricular fibrillation may occur in children diagnosed with WPW syndrome. Digitalis and verapamil are contraindicated in these patients because these drugs further slow the conduction of impulses in the AV node and enhance conduction in the accessory pathway.

Atrial Fibrillation in Wolff-Parkinson-White Syndrome ECG 36
The ECG above (figure 6) represents atrial fibrillation in WPW syndrome. There is a broad QRS complex tachycardia with a rate > 200 bpm, as well as an irregular rhythm with concomitant LBBB morphology (seen in V1). Although the ECG resembles atrial fibrillation with left bundle-branch, the other findings are not typical of LBBB, and the rate is too fast. Beat-to-beat variation in the QRS complex points to WPW syndrome. LBBB would show a consistent QRS complex.
Treating Atrial Fibrillation
Atrial fibrillation is treated pharmacologically, by ablation, or with a combination of both. Patients also undergo anticoagulation therapy for possible thrombus formation.
The clinician may also consider controlling the heart rate pharmacologically and treating the rhythm with electroconversion.

Chronic anticoagulants may treat AFib.
35 Cadogan M. Ashman phenomenon. Life in the fastlane website. Accessed May 22, 2019.
http://litfl.com/wp-content/uploads/2018/08/ECG-Ashman-aberrancy-5.jpg
36 Burns E. Preexcitation syndromes. Life in the fastlane website. Accessed September 4, 2020.
https://litfl.com/wp-content/uploads/2018/08/ECG-WPW-Atrial-fibrillation-3.jpg