Renin-Angiotensin-Aldosterone System (RAAS)
The renin-angiotensin-aldosterone system (RAAS) is a coordinated hormone pathway that helps your body regulate blood pressure, blood volume, and sodium and water balance. It activates when the kidneys sense reduced perfusion, low circulating volume, or reduced sodium delivery, and it responds by tightening blood vessels and increasing fluid retention to restore perfusion.
ACLS Certification Association videos have been peer-reviewed for medical accuracy by the ACA medical review board.
Article at a Glance
- The renin-angiotensin-aldosterone system (RAAS) helps the body maintain blood pressure and fluid balance.
- Generally, the RAAS response is desirable in hypovolemic patients and undesirable in patients with congestive heart failure.
- Clinicians will learn about the pathophysiology of RAAS and different drugs that work on the RAAS system.
What is the Renin-Angiotensin-Aldosterone System?
The RAAS is a hormone system that regulates blood pressure and fluid balance.
When the body has low blood volume or is hypotensive, the kidneys sense they’re not being perfused and release renin into the bloodstream, setting off a chain reaction that results in the release of angiotensin.

The renin-angiotensin-aldosterone system (RAAS) regulates blood pressure and fluid balance.
Angiotensin I is a hormone that gets converted to angiotensin II, a powerful vasoconstrictor. It elevates blood pressure and improves renal perfusion. Angiotensin II makes the blood vessels “tense” and causes vasoconstriction.
Next, aldosterone is released. Aldosterone is the “saltwater hormone.” When aldosterone is released, the body retains sodium and fluid while excreting potassium, increasing blood pressure and perfusing the kidneys.
Read: Human Immunodeficiency Virus (HIV)
Renin Angiotensin Aldosterone System (RAAS) Physiology
This video walks through the RAAS pathway step by step, showing how low renal perfusion triggers renin release, how angiotensin II increases blood pressure through vasoconstriction, and how aldosterone increases sodium and water retention. Watch the video to reinforce the sequence and the physiological purpose of each step.
What are the components of the RAAS and their functions?
Renin
Renin is an enzyme released by juxtaglomerular cells in the kidney when renal perfusion pressure drops, sodium delivery to the distal nephron decreases, or sympathetic stimulation increases. Renin initiates the cascade by converting angiotensinogen into angiotensin I.
Angiotensin
Angiotensin I is converted by angiotensin converting enzyme ace (commonly known as ACE), primarily in the lungs and vascular endothelium, into angiotensin II. Angiotensin II is a potent vasoconstrictor that raises systemic vascular resistance and supports blood pressure. It also stimulates aldosterone release, preferentially constricts the efferent arterioles to help maintain the glomerular filtration rate, and contributes to fluid retention and vascular remodeling.
Aldosterone
Aldosterone is released from the adrenal cortex and acts mainly in the distal convoluted tubules and collecting ducts of the kidneys. It promotes sodium and water reabsorption into the peritubular capillaries while increasing potassium excretion, and its dysfunction is central to adrenal disorders such as Addison’s vs Cushing’s. This expands intravascular volume and can increase blood pressure, which helps restore perfusion but can worsen fluid-overloaded states like CHF.
What organs and body systems are involved in the RAAS?
Kidneys
The kidneys act as the sensor and the trigger. They detect reduced perfusion, reduced sodium delivery, and increased sympathetic activity, then release renin to begin the RAAS cascade. The kidneys are also the main target organ for aldosterone-driven sodium and water reabsorption.
Liver
The liver produces angiotensinogen, the precursor protein that renin converts into angiotensin I. Without angiotensinogen, the RAAS cascade cannot proceed.
Lungs and Vascular Endothelium
Angiotensin converting enzyme (ACE) is found in high concentrations in lung tissue and along the vascular endothelium. ACE converts angiotensin I into angiotensin II, the primary effector hormone of the system.
Adrenal Glands
The adrenal cortex releases aldosterone in response to angiotensin II and other signals. Aldosterone directs the kidneys to retain sodium and water and excrete potassium, expanding circulating volume.
Hypothalamus
The hypothalamus contributes to thirst and fluid-seeking behavior and supports neurohormonal responses that help the body maintain perfusion during low-volume states.
Pituitary Gland
The posterior pituitary releases antidiuretic hormone (ADH), which increases water reabsorption in the kidneys. RAAS activation and angiotensin II can support ADH release, reinforcing water retention when blood volume is low.
How is the RAAS regulated?
RAAS activation begins when the kidneys detect reduced effective circulating volume. The most common triggers include low renal perfusion pressure, reduced sodium delivery to the macula densa, and increased sympathetic stimulation. These signals increase renin release, which drives the downstream formation of angiotensin II and aldosterone.
- Low renal perfusion: reduced renal blood flow and pressure in the afferent arterioles stimulates renin release
- Low sodium delivery: macula densa signaling increases renin when distal tubular sodium is reduced
- Sympathetic activation: beta 1 stimulation increases renin release during stress or hypotension
Counter-regulation: the body also has mechanisms that reduce RAAS activity when volume and pressure are restored. Atrial natriuretic peptide (ANP) and B type natriuretic peptide (BNP) promote vasodilation and sodium excretion and oppose aldosterone effects. As renal perfusion improves, negative feedback mechanisms act to inhibit renin release, helping prevent excessive vasoconstriction and fluid retention.
RAAS in Congestive Heart Failure
RAAS may adversely affect cardiac patients with congestive heart failure (CHF). Their heart isn’t pumping efficiently, or it’s lost some of its contractility. The kidneys aren’t adequately perfused, so they activate the RAAS system.
RAAS Activation in CHF: Quick Flow
- Reduced cardiac output leads to reduced renal perfusion
- Kidneys release renin, which increases angiotensin II
- Angiotensin II causes vasoconstriction and increases afterload
- Aldosterone and ADH increase sodium and water retention
- Result: fluid overload and higher cardiac workload, worsening CHF symptoms
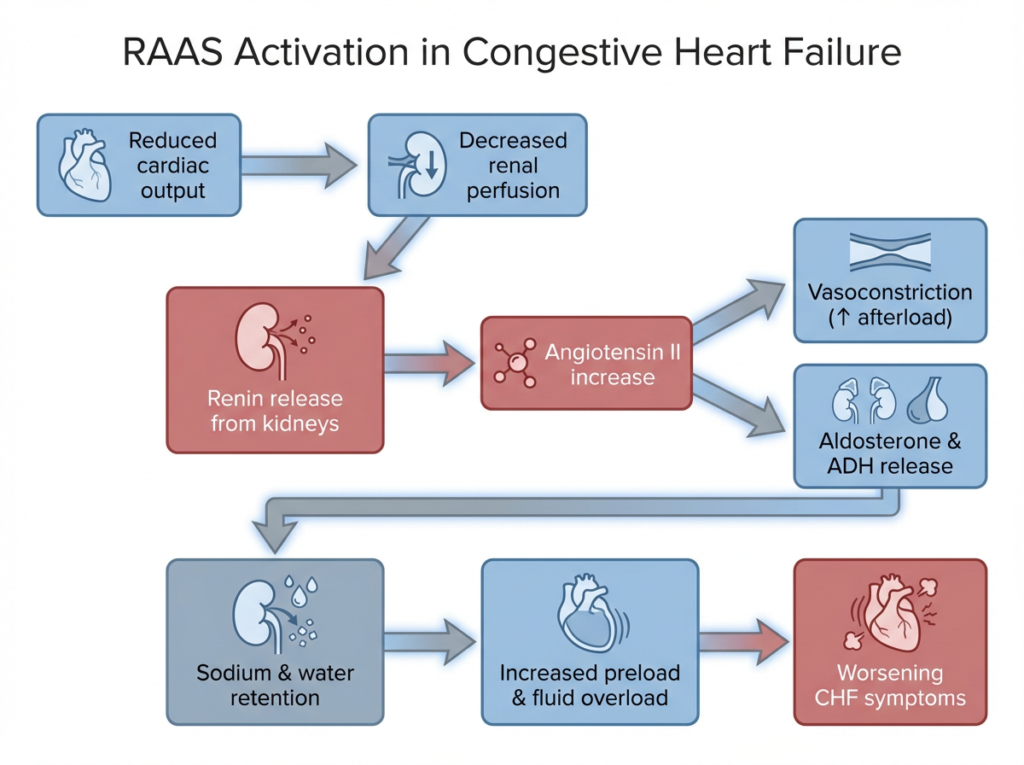
RAAS activation in congestive heart failure forms a maladaptive cycle that increases afterload and fluid retention, worsening cardiac workload.
In congestive heart failure, reduced cardiac output lowers renal perfusion, which the kidneys interpret as volume depletion. This triggers RAAS activation, leading to vasoconstriction and sodium and water retention. While initially compensatory, sustained RAAS activation increases afterload and preload, placing additional stress on the failing heart and worsening symptoms.
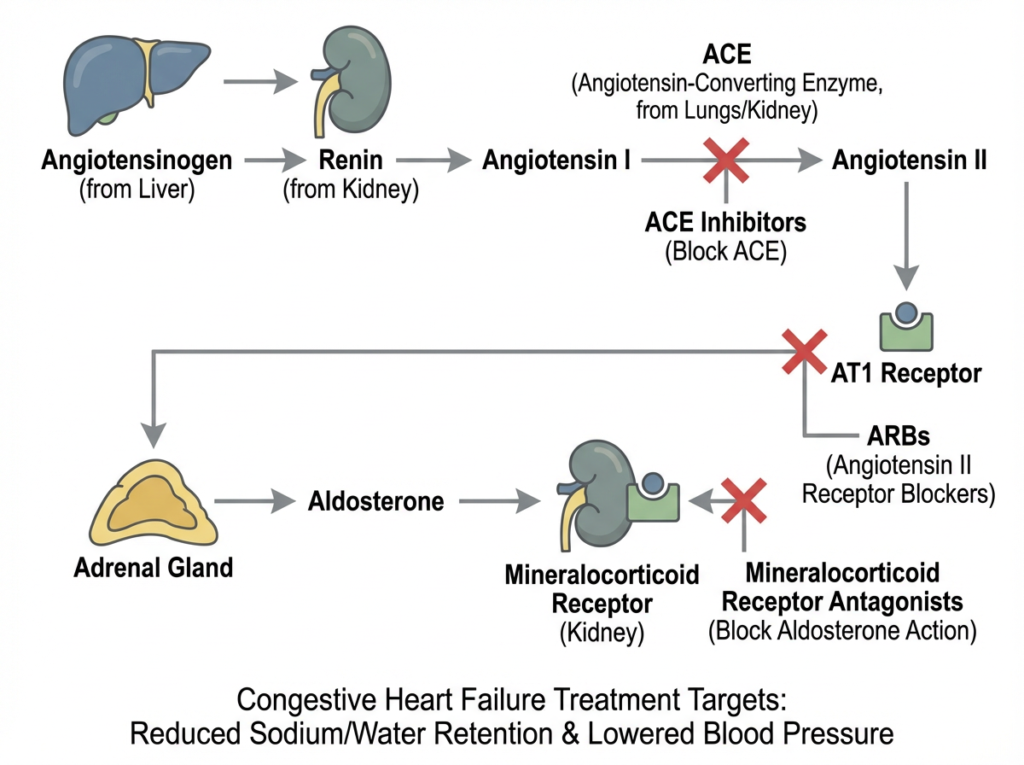
Common CHF medications target different points in the RAAS pathway to reduce vasoconstriction and fluid retention.
Many heart failure medications are designed to interrupt the RAAS pathway at specific points. ACE inhibitors reduce angiotensin II formation, ARBs block angiotensin II effects at the AT1 receptor, and mineralocorticoid receptor antagonists limit aldosterone driven sodium and water retention. Together, these therapies help reduce maladaptive neurohormonal activation in CHF.
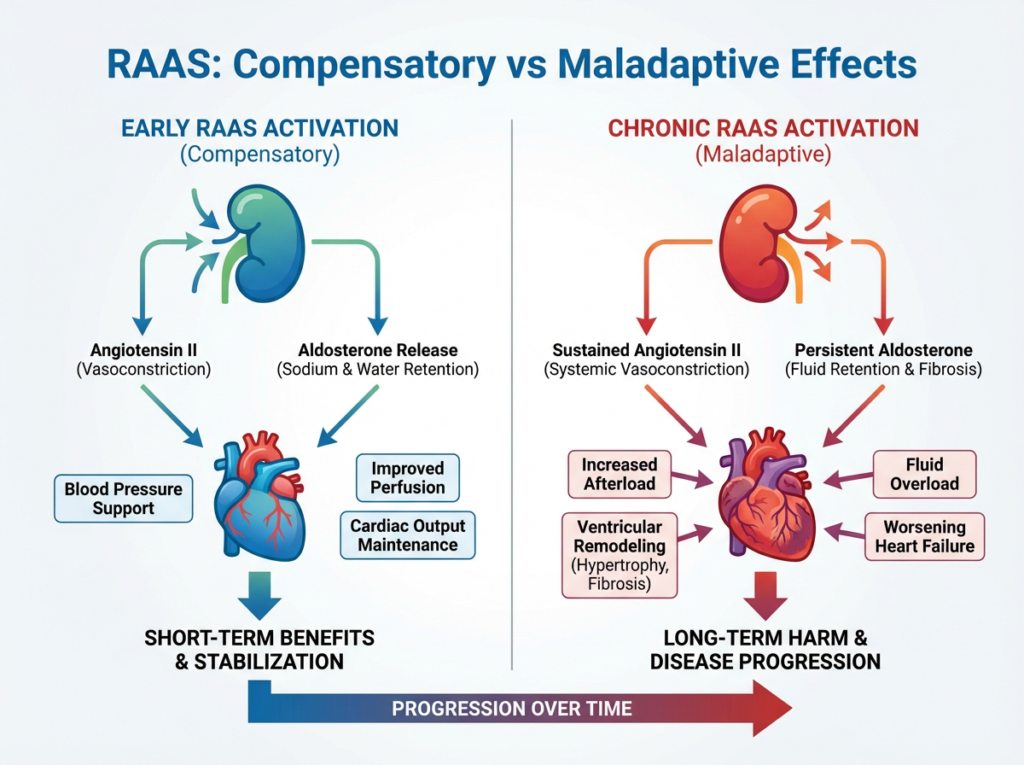
RAAS activation initially supports perfusion but becomes maladaptive when chronically activated in heart failure.
RAAS activation can be helpful in the short term by supporting blood pressure and organ perfusion. However, chronic activation leads to increased vascular resistance, fluid overload, and structural remodeling of the heart, contributing to progressive heart failure.

Activation of the renin-angiotensin-aldosterone system can worsen heart failure.
The active RAAS system causes vasoconstriction and retention of sodium and water, which is bad for CHF patients. They don’t need more fluids. Providers should administer angiotensin-converting enzyme inhibitors and ACE inhibitors to counter the reaction. They include the “-pril” drugs, such as lisinopril.
ACE-inhibitors block the angiotensin-converting enzyme and stop end reactions. Providers will see reduced vasoconstriction and sodium and increased fluid retention. Vasodilation will present, indicating the body now retains potassium instead of excreting it.

Lisinopril is an ACE-inhibitor. ACE-inhibitors block the renin-angiotensin-aldosterone system.
Angiotensin receptor blockers (ARBs) are similar to ACE inhibitors. Along with ACE inhibitors, they carry the risk of hyperkalemia due to the anti-aldosterone effects. Additionally, ACE inhibitors affect the bradykinin in the lungs, causing a dry cough. ARBs don’t affect bradykinin or present a dry cough risk.
Physicians also administer beta-blockers to treat RAAS in CHF patients. Beta-blockers improve ejection fractions, slowing the heart rate by blocking the sympathetic nervous system from stimulating the beta-receptors. While the heart rate is slowed, the ventricles have more time to fill, which is advantageous for CHF patients.
Providers should not administer beta-blockers to a patient in the acute phase of CHF. They reduce the heart’s contractility. If a patient is already hypotensive and bradycardic, reducing contractility and slowing the heart may worsen their condition.
The renin-angiotensin-aldosterone system maintains the body’s blood pressure and fluid balance. It works well for hypovolemic patients needing improved renal perfusion. However, it may adversely affect patients with CHF.
Angiotensin Receptors and CHF
Angiotensin II primarily acts through two receptor types that influence heart failure progression and medication targeting.
- AT1 receptor: drives vasoconstriction, stimulates aldosterone release, promotes sodium and water retention, and contributes to cardiac hypertrophy and remodeling. Many CHF therapies aim to reduce AT1 mediated effects.
- AT2 receptor: tends to support vasodilation and anti proliferative effects and may counterbalance some AT1 mediated actions in certain settings.
This is one reason ARBs are used in heart failure management: they block angiotensin II signaling at the AT1 receptor, reducing vasoconstriction and aldosterone driven fluid retention.
Pharmacological Targets in CHF
Several medication classes target RAAS and related neurohormonal pathways in CHF. The goal is to reduce maladaptive vasoconstriction, decrease fluid retention, and limit long term remodeling.
ACE Inhibitors
ACE inhibitors reduce conversion of angiotensin I to angiotensin II, which decreases vasoconstriction and reduces aldosterone signaling. They are commonly used long term, but require monitoring for blood pressure effects and potassium.
Angiotensin Receptor Blockers (ARBs)
ARBs block the AT1 receptor and reduce angiotensin II effects without increasing bradykinin, which is why they are less associated with dry cough compared with ACE inhibitors.
Mineralocorticoid Receptor Antagonists (MRAs)
MRAs such as spironolactone and eplerenone block aldosterone at its receptor. This reduces sodium retention and limits aldosterone mediated fibrosis and remodeling. Because MRAs can increase potassium, monitoring for hyperkalemia is important.
Beta Blockers
Beta blockers reduce sympathetic stimulation and can improve ejection fraction over time in stable chronic CHF. They are typically avoided in acute decompensated CHF until the patient is stabilized.
Clinical Implications of RAAS Dysfunction Beyond CHF
RAAS is clinically relevant beyond heart failure because chronic activation can contribute to persistent vasoconstriction, fluid retention, and progressive organ damage.
- Hypertension: excessive angiotensin II and aldosterone activity can increase systemic vascular resistance and promote sodium retention, sustaining high blood pressure.
- Chronic kidney disease: RAAS activation can worsen intraglomerular pressure and contribute to progressive kidney damage in some disease states.
- Diabetic nephropathy: RAAS modulation is commonly discussed in the context of protecting kidney function and reducing proteinuria under clinician guidance.
- Hyperaldosteronism: excessive aldosterone can lead to hypertension and low potassium, depending on underlying cause.
Because RAAS influences both vascular tone and renal handling of sodium and water, therapies that modulate RAAS are frequently considered across cardiovascular and renal conditions.
Frequently Asked Questions About RAAS
What is the main purpose of the renin angiotensin aldosterone system?
RAAS helps maintain blood pressure and effective circulating volume by increasing vasoconstriction and promoting sodium and water retention when renal perfusion is reduced.
What triggers renin release?
Renin release increases when renal perfusion pressure falls, sodium delivery to the distal tubule decreases, or sympathetic stimulation increases, especially through beta 1 receptors.
What is the difference between RAAS and the baroreceptor reflex?
The baroreceptor reflex is a rapid neural response that adjusts heart rate and vascular tone within seconds, while RAAS is a slower hormonal pathway that supports blood pressure and volume over minutes to hours through angiotensin II and aldosterone effects.
Why do ACE inhibitors sometimes cause a dry cough?
ACE inhibitors can increase bradykinin levels in the lungs, which may cause a persistent dry cough in some patients. ARBs are less associated with this effect because they do not increase bradykinin the same way.
More Free Resources to Keep You at Your Best
Editorial Note
ACLS Certification Association (ACA) uses only high-quality medical resources and peer-reviewed studies to support the facts within our articles. Explore our editorial process to learn how our content reflects clinical accuracy and the latest best practices in medicine. As an ACA Authorized Training Center, all content is reviewed for medical accuracy by the ACA Medical Review Board.
More to Learn
Fibrinolytic and endovascular therapies are the immediate therapies involved in stroke treatment. Read our detailed article on immediate therapies and their inclusion/exclusion criteria.
Gain insights into effective ACLS treatments for tension pneumothorax, a critical condition, by reading our article. Learn about life-saving interventions for this lung-related emergency.